
|
|
![]()
![]()
![]()
ONLY THE OVAL
BUTTONS LINK
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
|
Contents
Orthomolecular
Medicine |
Smart Drugs and Orthomolecular Substances for Aging Minds
by Richard P. Huemer, M.D. © 1999
Presented to the Advanced Longevity Workshop, ACAM, Nov. 18, 1998
Includes new text from Advanced Longevity Workshop, May 5, 1999
Journal of Advancement in Medicine 12(2):119-131, 1999
ABSTRACT. The aging of the nervous system is not fully understood, but defects in mitochondrial energy production, originating in mitochondrial DNA mutations, have been implicated, along with neurotransmitter deficiencies and excitotoxic mechanisms. Molecules involved in energy metabolism, such as NADH, acetyl-L-carnitine, ubiquinone, and lipoic acid, are potentially therapeutic. Various natural and synthetic pharmaceuticals can retard senility by influencing neurotransmitters, modulating NMDA-receptor mechanisms, enhancing circulation and metabolism, acting as antioxidants, and in other ways. Certain pharmaceuticals, including meclofenoxate, ergoloid mesylates, piracetam, ginkgo, and vinpocetine, have plural effects in the nervous system.
INTRODUCTION. Mankind is gifted with mental faculties far exceeding those of other creatures, but being human, we yearn to transcend the limits of Nature’s design. This drive to embellish upon Nature inspired the psychedelic movement of the 1960s, and to some extent it motivates the usage of so-called "smart drugs" today. Such usage in healthy people is, in my opinion, not medically defensible.
My reason for that statement is that most drugs possess undesirable pleiotrophic effects (which Bernard Rimland has called the "toximolecular principle"), so that long-term use of drugs may present health hazards. As a case in point, the Life Extension Foundation recently lowered its recommended intake of selegiline to only 10 mg per week following a report in the British Medical Journal of increased mortality among patients on long-term selegiline plus L-dopa. Moreover, some functions in aging systems are already highly upregulated; it is plausible that further stimulation might prove detrimental.
Smart drugs are those that improve cognitive functions, especially memory. Such functions decline with advancing age. Restoring failing cognitive functions is a legitimate use of smart drugs, and of what we might call "smart orthomolecular substances" as well. In the following paragraphs I will discuss several categories of smart drugs and orthomolecular substances, after first reviewing features of normal and pathological aging.
MECHANISMS OF AGING. Nobody knows with scientific certainty what causes aging, which is why theories of aging keep proliferating. Probably several of the theories will turn out to be correct, as mostly they are not mutually exclusive. Certainly the free-radical theory (1) explains a great deal, but it is not the entire story (2). The endocrine theory is relevant to vertebrate aging, but probably not to that of simpler creatures such as rotifers; it explains certain epiphenomena of aging but not the underlying process. Insufficient attention has been accorded to Strehler’s theory of genetic instability (3), probably because it promotes pessimism about any sort of effective intervention. According to Strehler’s evidence, certain kinds of redundant DNA, such as that which codes for ribosomal RNA, become progressively less redundant as the organism ages. It is easy to comprehend that when the last copy of ribosomal DNA has disappeared from a cell, protein synthesis on ribosomes will cease altogether (4).
Mitochondria have long been suspected of involvement in the aging process. The mitochondria from aged cells look distorted under the electron microscope, and according to many reports, they function poorly. Mitochondria are responsible for oxidative phosphorylation, the process that begins with the liberation of electrons from foodstuffs via the Krebs cycle. The electrons are passed by NADH (reduced co-enzyme form of niacin) to coenzyme Q10, and thence to protein complexes I-IV of the inner mitochondrial membrane, resulting in aerobic production of the master energy-molecule, ATP. Clearly, defective mitochondrial function will lead to deficient energy production, such as that observable in aged organisms.
MITOCHONDRIA AND DISEASE. Mitochondria are self-replicating organelles that contain their own DNA, which, like that of bacteria, is on a circular chromosome. The mitochondrial DNA (mtDNA) codes for 13 of the 60 polypeptides of the mitochondrial respiratory complexes; the others are products of the nuclear DNA. (Mitochondrial DNA also codes for mitochondrial ribosomal RNA and transfer RNAs.) Thus, nuclear and mitochondrial genetic systems are interdependent. This is a fortuitous arrangement, for if mitochondria were more self-reliant than they are, they might proliferate endlessly like intracellular disease germs.
I became interested in mitochondria many years ago, when I conducted studies on the half-life of mitochondrial DNA and membrane lipids in the brains of old and young mice. There were no differences in turnover rates of any of these constituents between young and old. However, the old mice had half the concentration of membrane lipids per unit weight of mitochondrial protein. Relative to the lipid constituents, the amount of DNA was about 40% greater in the old mitochondria. Moreover, I found through 3H-thymidine labelling that twice as much of the "old" mitochondrion’s DNA was newly synthesized. I also measured buoyant density of mtDNA--a crude way of screening for mutations--but found no differences (5).
Some researchers believe that mutations in mtDNA produce aging and disease by giving rise to dysfunctional mitochondria (6,7). The hypothesis is on shaky ground insofar as aging is concerned, since deletions and related lesions afflict less than 5% of the aging mtDNA, and typically an order of magnitude less than that. Indeed, intercellular nuclear- and mitochondrial-transfer experiments indicate that recessive nuclear-DNA mutations, not mitochondrial mutations, are responsible for decreased mitochondrial protein synthesis and cytochrome c activity in aged cells (8).
On the other hand, much higher levels of mitochondrial DNA damage are associated with certain diseases. Wallace (9) lists 12 mitochondrial DNA diseases and refers to the known or suspected existence of "dozens" of them. Most are quite rare, although Alzheimer’s and Parkinson’s diseases are all too common.
AGING DISEASES. In Parkinson’s disease a defect exists in the complex I enzyme NADH-CoQ10 oxidoreductase (or NADH reductase), in the substantia nigra and in other tissues as well. This implies a defective gene. The enzyme is encoded by both nuclear and mitochondrial genes, but cytoplasmic transfer experiments put the blame squarely on the mitochondrial genome (10). Interestingly, a Parkinson-inducing drug MPP+ (1-methyl-4-phenylpyridinium) inhibits the same complex I enzyme and also has the effect of diminishing by two-thirds the amount of mtDNA in cultured cells (11). Injectable NADH has been successfully employed to treat Parkinson’s disease. In a small prospective study, administration of 10 mg daily intravenously for 7 days was followed by statistically significant improvement in symptom scores, as well as significantly increased bioavailabity of plasma L-dopa (12).
In Alzheimer’s disease, point mutations have been found in the mitochondrial NADH dehydrogenase gene (13, 14) although not by all investigators (15). As in the case of Parkinson’s disease, NADH has been employed in a small open-label study as a treatment for Alzheimer’s disease. Following 8 to 12 weeks of therapy, all 17 subjects showed improved cognitive scores. The author cautioned that definitive conclusions cannot be drawn from this pilot study (16).
CoQ10 and alpha-lipoic acid would seem to have orthomolecular therapeutic potential in both Parkinson’s and Alzheimer’s disease, but apparently only CoQ10 has been studied clinically, in a small trial in parkinsonian patients, without notable effect (17). Lester Packer (18) has suggested that the metabolic co-factor and antioxidant lipoic acid might be beneficial in Alzheimer’s disease and other neurologic disorders, and indeed it has been proven to protect rodents’ brain cells from the ischemic damage that occurs when blood flow is interrupted (19), implying therapeutic value in stroke patients. Its potential benefit in Alzheimer’s disease has yet to be evaluated.
On the hypothesis that free radicals promote the brain deterioration of Alzheimer’s disease, a team at Columbia University conducted a 2-year comparison of the effects of vitamin E to those of selegiline, a monamine oxidase inhibitor. The vitamin E, in a daily dose of 2000 units, was somewhat superior to selegiline; it delayed the onset of severe Alzheimer’s disease by nearly 8 months compared with untreated patients, whereas the delay with selegiline was 7 months. The combination of the vitamin plus the drug was least efficacious (20).
One crucially important vitamin, namely B12, is deficient in the elderly more often than is commonly suspected, owing in part to the possibility of neural deficiency with normal hematologic findings. Its absence can promote dementia, and its replenishment can result in dramatically improved mentation and energy levels.
Another orthomolecular therapy for Alzheimer’s disease is acetyl-L-carnitine (ALC), a physiological molecule synthesized in mitochondria by the acetylation of carnitine, that functions in the transport of fatty acids into mitochondria to be oxidized for energy. Several hundred patients were involved in a double-blind study by Thal et al. (21). Patients with early-onset Alzheimer’s disease who took 3 grams of ALC daily deteriorated significantly less rapidly than did controls. (The late-onset treated patients fared slightly worse than controls, but not significantly so.) Of likely relevance is the fact that ALC can restore the damaged mtDNA transcription and translation in brains of aged laboratory rats (22). It also affects cholinergic metabolism and reduces lipofuscin accumulation, as do some of the smart drugs.
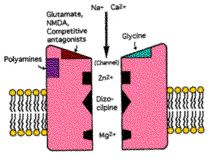
Interference with oxidative phosphorylation results in deficient ATP formation. From this follows partial neuronal depolarization, increased intracellular calcium and activation of NMDA receptors, with consequent excitotoxic cell death. High intracellular calcium can also lead indirectly to formation of the harmful peroxynitrite radical (23). In Alzheimer’s disease, the beta-amyloid protein found in neurofibrillary tangles and senile plaques is known to increase the vulnerability of neurons to excitotoxins such as glutamate and N-methyl-D-aspartate (NMDA)(24). Figure 1 depicts the potential interactions of the NMDA receptor.
|
Figure 1. Diagram of NMDA receptor in neuronal membrane, shown with binding sites and ligand-gated calcium channel (modified after Ingram et al., reference 29). |
|
SMART DRUGS. It is difficult to define exactly what a smart drug is, or even to define the more technical term nootropic drug (25). Some medical authorities, including the U.S. Food and Drug Administration, do not recognize the existence of this class of drug. Although there are many contenders for the designation of nootropic (listed by Dean et al. (26)), I will restrict the following discussion mainly to well-established drugs and their analogues, such as piracetam, meclofenoxate, selegiline, vincamine, and ergoloid mesylates. Much of the descriptive material that follows was extracted from the Life Extension Foundation’s Physician’s Guide to Life Extension Drugs (27).
A word about terminology is appropriate here. Many drugs go by several aliases, which may create the impression that many more drugs are available than actually exist. Table 1 lists some of the common synonyms.
Table 1. Synonyms for common nootropic and anti-aging drugs
|
Meclofenoxate |
Centrophenoxine |
Lucidryl |
|
Selegiline |
Deprenyl |
Eldepryl® |
|
Diethylamino ethanol |
DMAE |
Deanol |
|
Ergoloid mesylates |
Hydergine® |
. |
|
Antidiuretic hormone |
Vasopressin |
Diapid® |
Some of these substances have been known for quite a long time. Meclofenoxate, which is the dimethylamino ethyl ester of a chlorinated phenolic compound, was synthesized in 1959. It is listed in the 1968 Merck Index (28) as a medical analeptic and plant growth regulator. The same reference lists Hydergine as an adrenergic blocking agent, and Deanol as a CNS stimulant. The latter compound was first synthesized in 1904.
It is useful to classify such drugs by the predominant effect of each, since that is the therapeutic indication for the drug. (Some of the drugs have more than one major effect.) A classification for anti-Alzheimer’s strategies was suggested by Ingram et al. (29). Table 2 is rather loosely adapted from Ingram’s scheme, which was effect-based.
Table 2. Pharmacologic strategies for cognitive enhancement
|
Neurotransmitter-based |
|
•Acetylcholine - precursors, agonists, AChE inhibitors |
|
•Catecholamines - precursors, agonists, MAO inhibitors |
|
•Glutamate - NMDA agonists, polyamines, nitric oxide |
|
Circulatory enhancement: ginkgo, vinpocetine, KH3 |
|
Metabolic enhancement: piracetam, vinpocetine, Hydergine® |
|
Membrane modification: phosphatidyl serine |
|
Antitoxins: chelators (EDTA, desferoximine), calcium channel blockers |
|
Antioxidants: physiologic and pharmaceutical |
|
Anti-aptotic: flupirtine |
|
Miscellaneous: nerve growth factor; anti-amyloid agents; hormones; anti-crosslinking agents. |
NEUROTRANSMITTERS. The cholinergic system is relevant because of a deficit of cholinergic markers in the brains of Alzheimer’s patients, thought to be responsible for the loss of memory. Choline, citicholine, lecithin, DMAE, and meclofenoxate (because of its DMAE moiety) can all serve as acetylcholine precursors. Piracetam can increase acetylcholine synthesis.
Cholinergic agonists can be muscarinic, such as piracetam, or nicotinic, such as nicotine. Muscarinic receptors, as has long been known, can be blocked by scopolamine, which induces a profound temporary deficit in short-term memory. Interestingly, piracetam cannot reverse this short-term memory block but meclofenoxate can (30). Meclofenoxate is known to increase muscarinic binding affinity in the hippocampus and striatum (31).
![]()