
|
|
![]()
![]()
![]()
ONLY THE OVAL
BUTTONS LINK
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
J.
Theor. Biol. 67: 625-635, 1977;
reprinted
in Advances 1(3): 53-59, 1984.
A
Theory of Diagnosis for
Orthomolecular
Medicine
RICHARD P. HUEMER
Molecular Disease Institute, Agoura, California, U.S.A.
(Received 27 October 1975, and in revised form 18 June 1976)
SUMMARY
It is assumed that most diseases arise from multiple causes, and that
diseases
have the characteristics of polythetic classes. The signs and
symptoms
of clinically-apparent disease are epiphenomena, or emergent
properties
from the interaction among multiple biochemical etiologic
factors,
intrinsic and acquired. Each individual carries a unique set of
intrinsic
biochemical defects that are subsets of diseases to which he is
predisposed.
He acquires additional defects throughout life. Such bio-
chemical
defects can be detected by laboratory testing.
Clinically-apparent diseases are sets consisting of multiple laboratory-
test
anomalies associated with clinical signs and symptoms. Smaller sets
are
formed by laboratory-test anomalies pertaining to the functional state
of
major organ systems, without localizing signs and symptoms. The
latter
sets are termed preclinical disease. Small sets of laboratory-test
anomalies,
reflecting mainly intrinsic (genetic) defects, represent poten-
tial
disease. Under appropriate conditions, elements can be added to or
subtracted
from the sets, so that diseases may evolve to a more advan-ced stage
or regress under therapy. Ideally, sets of biochemical anomalies
should
be identified at an early stage, before expansion of the sets even-
tuates
in clinically-apparent disease.
1. Introduction
Orthomolecular medicine is defined as the provision of the optimum
molecular
constitution, especially the optimum concentration of substances
that are normally present in the body, for the purposes of treating
disease and preserving health (Pauling, 1968, 1974). Pauling's
innovative concept has had scant influence on the practice of
clinical medicine. Major impediments to acceptance of the
orthomolecular concept appear to be difficulties in identifying
molecular lesions and uncertainties over the meaning and measurement
of optimal. Consequently,
it is difficult to select appropriate therapies and monitor
therapeutic responses with confidence. In general, the few clinicians
who practice orthomolecular therapy tend to assume that the existence
of a given clinical syndrome implies a particular biochemical
pathosis, in much the same manner that wet streets imply rain. Thus
orthomolecular medical practice is hindered not only by diagnostic
imprecision , but also by a post hoc
fallacy that rests on traditional (and, it will be shown, antiquated)
diagnostic methodology.
The human organism
can be regarded as a set of subsystems, of which integrated
functional measurements can serve to predict the status of the
organism as a whole (Patton, Huemer, Hussman & Caines, 1963).
Along functional lines, subsystems may be identified as assimilatory,
excretory, regulatory and circulatory; or, according to a more
traditional schema, digestive, renal, neuroendocrine, cardiovascular,
etc. Such subsystems are further divisible into groups of cells
performing specialized functions characteristic of one (or more)
subsystems. In each group of cells is expressed that portion of the
organism's genome that is relevant to the specialized functions.
Differing groups of cells possess similarities as well as differences
among their complements of active genes, and thus possess
similarities as well as differences in their metabolic patterns. This
fairly conventional way of regarding the living organism can serve as
the departure point for novel insights into the nature of disease and
the method of its diagnosis.
2.
Nature of Disease
A disease is an
abnormal functional state characterized by a set of symptoms and
physical findings. Diseases as seen in individual patients often
depart from textbook descriptions of disease in that some of the
characteristics may be lacking, or the characteristics may be
associated with those of other disease states. Diseases possess the
characteristics of polythetic classes*. According to Sokal &
Sneath (1963), a polythetic class fulfills the following criteria:
(a) each member of the class possesses a large (but unspecified)
number of the properties that characterize the class as a whole; (b)
each property of the class is possessed by large numbers of the class
members. In addition, a class is deemed fully polythetic if no
property of the class is possessed by every member of the class.
___________________________
*The
term "fuzzy sets" has a similar meaning.
Disease classes are polythetic with respect to etiology as well as symptomatology. It is now generally understood that many--perhaps most--diseases result from multiple causes. A class such as tuberculosis is not fully polythetic because all cases possess a common etiologic factor (the tubercle bacillus) in addition to various other etioiogic factors. However, some of the chronic degenerative diseases may be fully polythetic with respect to etiology, which may explain why their control is elusive; there may exist no single point at which the process can be controlled. To restate the matter, the organism can become diseased for a variety of reasons, but there may be only a limited spectrum of possible dysfunctional responses to an immense number of possible combinations of causative factors.
Since
the living organism is a biochemical mechanism, the causes of dys-
function
must at their most fundamental level be biochemical in nature. In
the
final analysis, except for traumatic conditions, all diseases become
molecular,
whether the defects are congenital or acquired (Huemer, 1972).
Some
biochemical lesions are acquired during life when environmental
stresses
(e.g. toxins, malnutrition, infection) exceed the reparative
capacities of the living system. Others are intrinsic, or genetic. It
will be recalled that groups of cells possess differences from and
similarities to each other with respect to their biochemical patterns
and active-gene complements. A set of genetic defects might affect
one specialized group of cells more than others, but would probably
affect many cell groups to some degree. The polythetic arrangement of
gene-expression sets is echoed in the polythetic nature of disease itself.
3. Genetic Substrate of Disease
From
the outcome of consanguineous matings it appears that the average
human
being is heterozygous for one or two genes that would cause serious
disease
in the homozygote (Carter, 1967). Muller (1950) estimated that the
average
human is heterozygous for a minimum of eight, and possibly scores
of
genes, each of which produces a slight detrimental effect. According to
Muller,
the combination of such genes, acting together, gives the individual
his
own characteristic pattern of weakness. More recently, Hubby & Lewontin
(1966)
demonstrated a previously unsuspected degree of polymorphism
among
the genes of Drosophila pseudoobscura in wild populations; at least
39
% of the loci in the genome are polymorphic over the whole species, and
an
estimated 8 to 15% of an individual fly's genome is in the heterozygous
state.
If the finding may be generalized to humans--and there is no compelling
reason
to suppose otherwise--then Muller's estimate for prevalence of sub-
vital
genes seems conservatively low. Furthermore, any individual will be
homozygous
for numerous isoalleles that may confer reduced fitness only
in
the homozygous state.
Along
related lines, the enzyme studies of Harris (1966) led him to con-
clude
that enzyme polymorphism may be a fairly common phenomenon
among
the very large numbers of enzymes that exist in humans. The degree
to
which enzyme polymorphism affects health is not presently known. It
appears
quite probable, at any rate, that the average human individual
carries
thousands of isoalleles, and it is reasonable to suppose that many of
the
isoalleles are subvital to a degree. Thus the individual will carry through-
out
life a characteristic pattern of molecular weaknesses.
In
principle it should be demonstrable that an individual carries a unique
pattern
of biochemical anomalies that persists, with variable degrees of
expression,
for many years. The available data on normal characters (Cotlove,
Harris
& Williams, 1970) indicate that an individual's biochemical measure-
ments
generally remain quite stable over a span of many months, fluctuating
narrowly
about homeostatic set-points. A similar study seems not to have
been
conducted on abnormal characters, but this author has observed a
27-year-old
Caucasian male who had fasting hyperg]ycemia, a-lipopro-
teinemia,
relative lymphocytosis, high salivary uric acid, high salivary
cholesterol,
and high salivary chloride; salivary ascorbic acid was abnormally
low.
The patient had previously been examined at the ages of 12, 14 and 16
years.
He had a relative lymphocytosis on all three occasions, low salivary
ascorbic
acid on two occasions (borderline-low on the third), and high
salivary
cholesterol on two occasions; high salivary chloride and borderline-
high
fasting glucose had been found on the single prior occasion when those
tests
had been performed. Relevant to a possible genetic origin of the
anomalies,
it should be noted that all of the findings except fasting hyper-
glycemia
were present in one or both parents, and one parent has an aberrant
glucose-tolerance
curve.
4. Classification of Disease
The
objection might be raised, in relation to the referenced clinical-
laboratory
studies, that the tests provide only indirect measurements of
genetic
qualities, and probably reflect polygenic traits; hence they do not
necessarily
indicate potential molecular causes of any disease but instead may
be
effects of underlying genetic pathology. Furthermore, since most clinical-
laboratory
measurements are made under homeostatic conditions, they will
fail
to detect numerous weaknesses (reduced capacity for enzyme synthesis,
for
example) for which the organism is able to compensate in the unstressed
state.
The objections are valid in so far as they apply to any attempted use
of
such data to classify disease along strictly etiologic lines. At
present, it is
not
feasible to measure large numbers of direct gene-products in the intact
organism;
hence a strictly molecular-etiologic classification is not presently
possible.
However,
it is not necessary to know cause-and-effect in order to classify
diseases
by biochemical and other laboratory-test criteria. It is not necessary,
for
example, to know whether hyperuricemia originates in a hereditary
enzymic
defect or as the result of ingestion of drugs or excessive beef, in
order
to state that hyperuricemia belongs to the class, gouty arthritis.
It is
not
necessary to explain the absence of hyperuricemia in some cases of gout
or
its presence in other diseases, in order to place hyperuricemia in
the gouty-
arthritis
class. For purposes of disease classification, laboratory-test anomalies
possess
the same significance as clinical signs and symptoms, with this
important
difference: laboratory-test anomalies are subsets of signs and
symptoms,
and are less far removed than clinical signs from the ultimate
molecular
causes.
Sokal
& Sneath, in suggesting the application of polythetic concepts to
the
construction of disease taxa, have commented that etiology may be
unsuitable
as a general principle for defining taxa (Sokal & Sneath, 1963,
p.
283). However, a truly etiologic classification is theoretically
possible and
will
surely be achieved in time; meanwhile, a polythetic biochemical-phenetic
classification
should serve as a useful approximation to that ideal.
A
few attempts have been made at mathematical diagnostic interpretation
from
multiphasic biochemical test data. Thus, Pomeroy (1975) reports
statistically
significant separation of hamsters into original vendors' groups
by
means of computerized linear discriminant analysis of plasma amino acid
profiles.
Using computerized pattern-recognition calculations, Robinson &
Pauling
(1974) have demonstrated correlation of chromatogram patterns
with
sex, fasting, birth-control medication and neuromuscular disorders. A
criticism
of such work is that the experiments are usually not based on
known
biochemical mechanisms related to particular diseases, so that it is
difficult
to reason inductively from the data. By ignoring mechanisms and
limiting
the methodology to traditional, clinically-established diagnostic
categories,
one invites difficulties in dealing with undifferentiated early
disease,
transitional states, semantically imprecise categories, and multiple
simultaneous
diseases.
Implicit
in the preceding discussion is the assumption that normality can
be
defined; otherwise, anomalous has no meaning. Many physicians believe
that
each individual possesses his own "normal" set of
biochemical test
values,
by which it is meant that the individual's health is good when various
tests
give certain numerical results, whether or not those results fall within
the
statistically normal range. I reject this arbitrarily relativistic view.
Normality,
in the context of this discussion, means normally distributed
about
a mean; in clinical laboratory medicine, the normal range of a para-
meter
is plus or minus two or three standard deviations from the mean of a
healthy
population (Amador, 1975; Copeland, 1972; Files, Van Peenen &
Lindberg,
1968). Strictly speaking, the distribution of test values in healthy
persons
is not Gaussian, as Elveback, Cuillier & Keating (1970) point out,
and
the false assumption of a Gaussian distribution can lead to mis-
classification
of some test results as "normal". Regardless of the chosen
statistical
criteria, it seems clear that most or all of the "normal range"
corresponds
to that condition which is termed by geneticists the "wild type",
symbolized
by +. Deviations from the wild type are not manifestations of
individual
good health, but of single or multiple genetic defects (including
associated
compensatory mechanisms). Cheraskin & Ringsdorf (1973)
analyzed
the mean and variance of fasting blood-glucose values for 100
dental
patients. By selecting the patients within this group according to
progressively
more strict criteria for oral health, they achieved, by stages, a
slight
reduction in the mean and a tenfold restriction in the normal range.
It
is reasonable to suppose that laboratory-test values should be standardized
on
populations meeting only the most stringent criteria for health and vigor
(cf.
Files et al., 1968; Cheraskin & Ringsdorf, 1973).
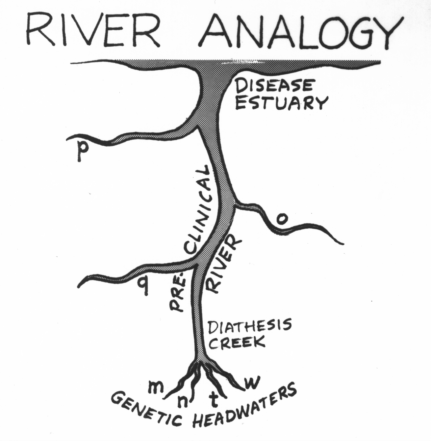
5. Evolution of Disease
Clinically-apparent disease usually has evolved through stages of potential
disease
and preclinical disease, the latter being a dysfunctional state of one
or
more major subsystems that does not produce a characteristic set of
symptoms
and signs. In the beginning exist isolated biochemical anomalies
(diatheses),
representing the individual's unique genetic pattern. Each
diathesis
may be regarded as a potential disease. As further anomalies
accumulate
(resulting from the interaction of environmental stresses and
constitutional
weaknesses), the stage of preclinical disease is attained. This
tendency
to acquire biochemical anomalies with advancing age shows up
as
an age-related broadening of distribution curves for laboratory test
values,
as observed by Files et al. (1968). Possible mechanisms include exo-
genous
agents (toxins, viruses), age-related enzyme polymorphism (Gershon
&
Gershon, 1973), and failure of homeostatic feedback loops (as in the post-
menopausal
deregulation of FSH secretion). Finally, the accumulation of
still
more anomalies results in frank symptoms and physical findings. To
put
the matter in symbolic terms, let X be a polythetic class (disease)
characterized
by the anomalies m, n, o, p and q. At time T0
the individual
possesses
m and n, plus the unrelated anomalies t and w.
As mntw do not
form
the major part of a disease set, the individual is apparently healthy.
At
T1, q is added to the individual's set. The individual
at this point may feel
vaguely
ill, as more of the set mnopq has been completed, and q may perhaps
be
part of another set that has also been brought nearer to completion.
Finally,
at Tx, o and/or p occurs, and the individual
experiences a case of X.
![]()
![]()