
|
|
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
|
PAGE 2 |
J.
Theor. Biol. 67: 625-635, 1977; |
A Theory of Diagnosis for Orthomolecular Medicine
5. Evolution of Disease (continued)
Consider the following scenario, in which disease X is
characterized by
abcde,
disease Y by defgh, and disease Z by dhjkm.
A patient possesses in-
herent
anomalies dfm, and is seemingly healthy. After a period of
stress and
poor
diet, anomaly e develops. The patient now has a sufficiently
large set
(defm)
to experience vague constitutional symptoms such as fatigue and
headaches.
He consults his physician, who is uneasily aware that most
patients'
complaints do not resemble the textbook syndromes that he has
studied
in the teaching hospital. The physician does not perform a battery
of
tests, as the health insurance will not pay for them; the physician
in any
event
has not been properly trained to interpret the set defm. The physician
does
make a provisional diagnosis of Y-or-Z, for he knows
that both diseases
are
often associated in their early stages with vague constitutional symptoms.
To
clarify the diagnosis he tests for dgm. Finding dm, he
assumes Z and
erroneously
excludes Y, which has actually evolved most closely toward a
clinical-disease
stage. He prescribes a drug that has the unfortunate side-
effect
of activating c. Additional symptoms arise, of an ambiguous nature;
the
patient is now chronically ill and the prolonged derangement in body
chemistry
finally causes compensatory mechanism a to fail. The distraught
patient
then consults a university medical school professor, who perceives
from
the symptoms that the differential diagnosis lies between X
and Y.
Testing
for abcdefgh, and finding acdef, the professor
concludes that X is
the
correct diagnosis, Y is unlikely, and Z is erroneous.
This ritual, repeated
endlessly
in medical centers throughout the world, serves to affirm the natural
order
of a universe in which professors of medicine possess more erudition
than
their brethren in private practice. In fact Y (in a
preclinical stage) has
been
neglected, and Z (in a potential stage) ignored altogether.
If by chance
a
test for m should be performed, the professor may publish a
clinical note
on
the unusual occurrence of m in a case of X.
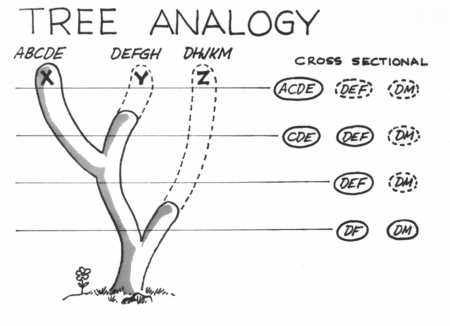
The point of the preceding scenario is that diseases progress through
evolutionary
stages, that several evolving diseases are likely to be present
simultaneously,
that differential diagnosis is therefore a pointless exercise,
and
that insufficient data lead to inadequate conclusions.
It is presently exceptional for disease to be recognized in its preclinical
stages,
and indeed no unifying scheme exists for classifying preclinical
diseases.
A suitable scheme might be classification according to subsystem
(cardiovascular,
adrenal, hepatic, etc.) and process (e.g. insufficiency,
imbalance,
hyperfunction, congestion). Thus a hierarchy is established in
which
constitutional abnormalities combine with acquired anomalies to
produce
derangements of subsystems; interactions among abnormally
functioning
subsystems result in clinically-apparent diseases. For example, a
patient
with elevated excretion of metanephrines and pregnandiol, with low
17-ketosteroids
and cortisol, may be said to have adrenal imbalance. Later,
when
subsequent exhaustion of the adrenal glands leads to widespread
systemic
disturbances, he can be diagnosed as having Addison's disease.
The
clinically-apparent disease diabetes mellitus may be preceded by dys-
functional
states of the pancreas, adrenals, pituitary, thyroid and peripheral
vasculature,
as discussed by Loeb (1955) or by Medalie, Papier, Goldbourt
&
Herman (1975). The latter authors used a multivariate analytical tech-
nique
to demonstrate that serum uric acid and cholesterol, among other
parameters,
are major factors in the development of diabetes.
In
practice, a patient will usually be found to have several preclinical
diseases
at the same time. Examples of preclinical diseases with their com-
ponent
anomalies are presented in Table 1. Such sets of anomalies would
be
encountered fairly frequently were it not for inadequate laboratory
examinations
that test for only a few disease elements among the many
within
a set, an approach that is insufficiently multivariate. Unfortunately,
the
selected elements are most often those that differentiate the hospitalized
population
from the outpatients.
TABLE
1. Diagnostic profile of a 40-year-old male physician
who
complained of chronic fatigue
|
. |
Hepatic insufficiency
Malabsorption dysnutrition syndrome
Adrenal hyperfunction
Pancreatic dysfunction Hypogammaglobulinemia Reticulocytopenia |
. |
Diagnostic
categorical headings are followed by a list of the component anomalies.
The
final two diagnoses are isolated anomalies that do not yet form part
of a larger set. The data were obtained by standard biochemical and
cellular analyses of body fluids.
6. Therapeutic Implications
Although
a discussion of orthomolecular therapy (Pauling, 1974) is
beyond
the scope of this paper, it is appropriate to mention that many of
the
biochemical anomalies within a disease set are in theory reversible by
orthomolecular
methods. Preclinical disease ought to be more easily reversible
than
clinically-apparent disease, because preclinical-disease sets are smaller
than
clinical-disease sets. For example, examination of an infant's urine
may
reveal the diathesis phenylketonuria. Once recognized, the potential
disease
can be treated by orthomolecular methods, thereby preventing the
clinically-apparent
disease phenylpyruvic oligophrenia, of which phenyl-
ketonuria
is a subset. As an additional example, I have observed a
68-year-old
woman who was treated for multiple clinical and preclinical
diseases,
including chronic hepatic dysfunction. The latter condition was
defined
by high serum copper, high cholesterol esters, high isocitric de-
hydrogenase,
low cholinesterase and low thermostable alkaline phosphatase
isoenzyme.
The patient responded to orthomolecular therapy with a reversion
to
normal of the anomalous esterified cholesterol, isocitric dehydrogenase
and
cholinesterase. Presumably, if this patient were subsequently to be
stressed
with a hepatotoxin, the now-latent anomalies would appear more
readily
than would unrelated anomalies.
7. Conclusions
The art of clinical diagnosis conventionally rests on the assumption that
signs,
symptoms and anomalous laboratory findings follow from specific
diseases,
each of which in turn follows from unique causes. By contrast, the
theory
presented here holds that multiple non-specific causes give rise first
to
biochemical aberrations, which then result in symptoms and signs, from
which
a perceived disease state follows. The present theory thus constitutes
an
inversion of the usual conceptual hierarchy.
The present model is more deterministic than the conventional one, in
that
a more central role is assigned to genetic influences. The model predicts
that
unique sets of stable biochemical anomalies will be observable in each
individual,
regardless of his stage of development. It predicts further that
some
elements of such sets will become observable as components of the
diseases
that an individual will develop with the passage of time. Additionally,
the
model requires that disease states as conventionally defined be describable
as
polythetic classes of biochemical anomalies.
If the predictions of the theory are valid, the science of clinical diagnosis
should
become a process of multifactorial biochemical analysis, followed by
assignment
of biochemical anomalies (with probability weightings) to various
diagnostic
categories. By this means it will be possible to identify evolving
diseases
in early (preclinical) stages of development, or even as mere
genetically-determined
predispositions. The diagnostic process would appear
to
lend itself well to electronic data-processing techniques (cf.
Pomeroy et al.,
1975).
In the final analysis, most diseases are traceable to molecular causes,
whether
inborn or acquired during life. Present-day clinical-laboratory
technology
is sufficiently advanced to detect the proximate effects of many
molecular
defects, and in some cases the defects themselves. Thus the
classification
of disease along the lines elaborated above is presently feasible.
In
time, refinements in laboratory technology will permit
identification of a
sufficiently
broad range of molecular defects to make possible a true
molecular-etiologic
classification of disease.
The theoretical analysis implies that laboratory tests should
properly be
used
to make a diagnosis before a patient becomes ill, not afterward. The
timely
application of specific therapy can then prevent more serious disease.
Isolated
biochemical defects can in principle be corrected with orthomolecular
methods,
and a small number of defects should be more easily correctible
than
a larger number. At present it is indifference, not technological im-
maturity,
that offers the major obstacle to earlier disease detection and to
advanced
preventive therapeutics.
I
am profoundly grateful to the late Joseph D. Walters, M.D., for many dis-
cussions
concerning the diagnostic interpretation of his clinical data. These pages
contain
descriptions of clinical cases observed by Dr. Walters and myself. I also
acknowledge
the helpful comments of Dr. Bernard Strehler and Dr. Leon Pomeroy
on
earlier versions of this manuscript. I thank Damon Medical
Laboratory, Sherman
Oaks,
California, for donating the tests on which Table 1 is based.
REFERENCES
AMADOR, E. (1975). J. Am. med. Assoc. 232, 953.
CARTER, C. 0. (1967). Lancet i, 436.
CHERASKIN,
E. & RINGSDORF, W. M. (1973). Predictive Medicine: A Study in Strategy
p. 65. Mountain View: Pacific Press.
COPELAND,
B. E. (1972). In Clinical Diagnosis by Laboratory Methods (I.
Davidsohn &
J. B. Henry, eds), p. 1. Philadelphia: W. B. Saunders.
COTLOVE, E., HARRIS, E. K. & WILLIAMS, G. Z. (1970). Clin. Chem., 16, 1028.
ELVEBACK, L. R., GUILLIER, C. L. & KEATING, F. R. (1970). J. Am. med. Assoc. 211, 69.
FILES, J. B., VAN PEENEN, H. J. & LINDBERG, D. A. B. (1968). J. Am. med. Assoc. 205, 94.
GERSHON, H. & GERSHON, D. (1973). Mech. Age. Dev., 2, 33.
HARRIS, H. (1966). Proc. R. Soc. B. 164, 298.
HUBBY, J. L. & LEWONTIN, R. C. (1966). Genetics, Princeton 54, 595. [excuse my typographical error]
HUEMER, R. P. (1972). J. appl. Nutr. 24, 34.
LOEB,
R. F. (1955). In A textbook of Medicine (R. L. Cecil & R. F.
Loeb, eds), p. 658.
Philadelphia:
W. B. Saunders.
MEDALIE,
J. H., PAPIER, C. M., GOLDBOURT, U. & HERMAN, J. B. (1975).
Arch. intern.
Med.
135, 811.
MULLER, H. J. (1950). Am. J. hum. Genet. 2, 111.
PATTON, D., HUEMER, R. P., HUSSMAN, T. A. & CAINES, K. L. (1963). Advanced Psycho-physiological Sensors for the PIAPACS Program. Los Angeles: Planning Research Corp. (Report R-449).
PAULING, L. (1968). Science, N. Y. 160, 265.
PAULING, L. (1974). J. int. Acad. prev. Med. 1, 1.
POMEROY, L., KEYES, J. & PATTERSON, J. (1975). J. int. Acad. prev. Med. 2, 53.
ROBINSON, A. B. & PAULING, L. (1974). Clin. Chem. 20, 961.
SOKAL,
R. R. & SNEATH, P. H. A. (1963). Principles of Numerical
Taxonomy, p. 14. San
Francisco:
Freeman.
![]()